Bayesian Linear Regression
By now we know what a linear regression looks like. Let's consider a special case where number of parameters, p = 0: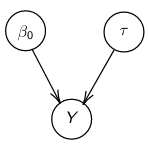
Assume Y is distributed as a normal distribution with mean β0:
Y|β0, τ ∼ N(β0, σ2 = 1/τ )
τ = 1 / σ2 , also called precision <- Be aware this will be used interchangeably with variance
The density function is: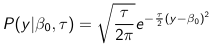
Mean and variance:
E(Y) = μ0; V(Y) = 1/τ0 + τ
The Posterior Distribution for β0 is calculated using Bayes' Theorem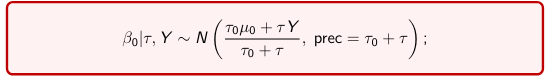
When Mean and Variance are Unknown
We use a Normal prior distribution for the mean β0 ∼ N(μ0, τ0) and a Gamma prior distribution for the precision parameter: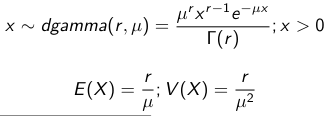
JAGS example:
model.1 <- "model {
for (i in 1:N) {
hbf[i] ~ dnorm(b.0,tau.t)
}
## prior on precision parameters
tau.t ~ dgamma(1,1);
### prior on mean given precision
mu.0 <- 5
tau.0 <- 0.44
b.0 ~ dnorm(mu.0, tau.0);
### prediction
hbf.new ~ dnorm(b.0,tau.t)
pred <- step(hbf.new-20) # hbf >= 20
}"Predictive Distributions
Given the Prior and Observed data we can compute the probability of a new observation will be greater or less than some integer threshold. The predictive distribution is a distribution of unobserved y~, that is: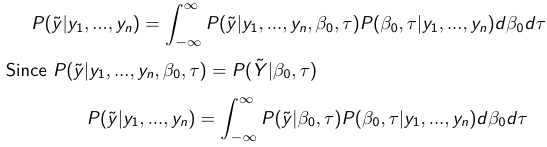
The two sources of variability in prediction are in the parameters V(β0, τ | y) and the variability in the new observation V(y | β0, τ)
- To simulate from predictive density, do repeatedly:
- Sample one sample β0*, τ* from posterior β0, τ | y
- Sample one y~ ~ N(β0*, 1/τ* )
- During the Gibbs sampling we generate samples values from the posterior distribution β0, τ | y
- So Generating y~ ~ N(β0, τ | y) will produce the correct predictive distribution samples. P(y~ > 20 | data) is the proportion of y~ > 20
Log Normal Distributions
Since in the example above the outcome distribution can only be positive we can use a Log-Normal distribution, a continuous distribution with support for values y > 0.
Y | η, σ2 ~ INormal(η, σ2) with density function: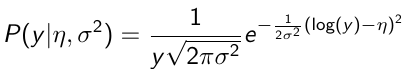
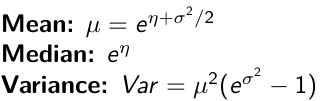
It also implies: Log( Y )~ Normal(η, σ2)
In JAGS we use: dlnorm(η, σ2)
"model {
for (i in 1:N) {
hbf[i] ~ dlnorm(lb.0,tau.t)
}
## prior on precision parameters
tau.t ~ dgamma(1,1);
### prior on mean
lb.0 ~ dnorm(1.6,1.6);
b.0 <- exp(lb.0)
### prediction
hbf.new ~ dlnorm(lb.0,tau.t)
pred <- step(hbf.new-20)
}"In the above example, we get the initial prior on β0 from previous data, where we derived median(b.0)=5 and V(b.0)=1/0.44=2.27
Then we take the log of the median to get η and transform the precision variable with: log(1 + V (b.0)/E (b.0)2 ), then take the inverse of that to get the variance.
Parameter Interpretation
Let's consider data of the SNP rs766432 on the effect of HbF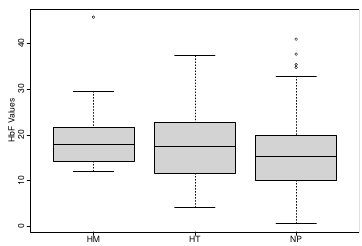
Where HT is heterzygote, HM is homozygoute and NP is common alleles. 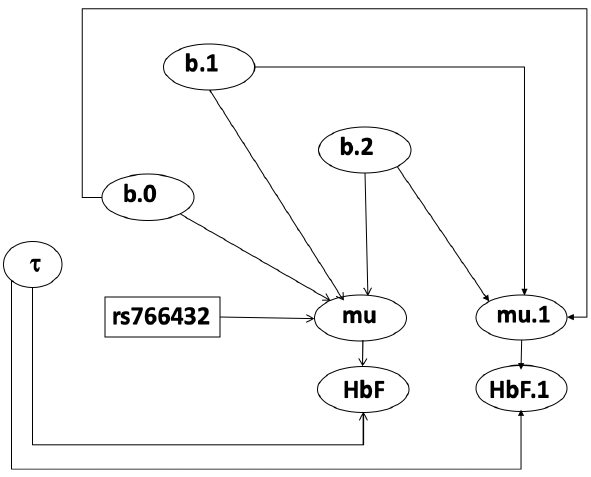
- We start by creating indicators for HT and NP using the equals( , ) function in JAGS
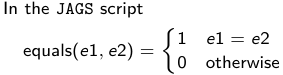
- The mean of Log HbF
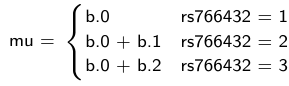
b.1 = the effect of HT vs HM
b.2 = the effect of NP vs HM - The hypotheses:
1. H0: b.1 = 0; HM and HT have the same effect
2. H0: b.2 = 0; HM and NP have the same effect
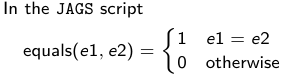
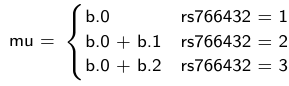
model.1 <- "model {
for (i in 1:N) {
hbf[i] ~ dlnorm(mu[i],tau.t)
mu[i] <- b.0+b.1 *equals(rs766432[i],2)+b.2 *equals(rs766432[i],3)
}
## prior on precision parameters
tau.t ~ dgamma(1,1);
### prior on mean given precision
b.0 ~ dnorm(0, 0.001);
b.1 ~ dnorm(0, 0.001);
b.2 ~ dnorm(0, 0.001);
### prediction
lmu.1 <- b.0;
hbf.1 ~ dlnorm( lmu.1,tau.t);
pred.1 <- step(hbf.1-20)
lmu.2 <- b.0+b.1;
hbf.2 ~ dlnorm( lmu.2,tau.t);
pred.2 <- step(hbf.2-20)
lmu.3 <- b.0+b.2;
hbf.3 ~ dlnorm( lmu.3,tau.t);
pred.3 <- step(hbf.3-20)
### fitted medians by genotypes
mu.1 <- exp(lmu.1)
mu.2 <- exp(lmu.2)
mu.3 <- exp(lmu.3)
par.b[1] <- b.0;
qpar.b[2] <- b.1;
par.b[3] <- b.2
par.h[1] <- hbf.1;
par.h[2] <- hbf.2;
par.h[3] <- hbf.3;
par.m[1] <- mu.1;
par.m[2] <- mu.2;
par.m[3] <- mu.3
par.p[1] <- pred.1;
par.p[2] <- pred.2;
par.p[3] <- pred.3
}"
data.1 <- source("saudi.data.2.txt")[[1]]
model_hbf <- jags.model(textConnection(model.1), data = data.1,n.adapt = 1000)
update(model_hbf, 10000)
test_hbf <- coda.samples(model_hbf, c("par.b", "par.h", "par.m","par.p"), n.iter = 10000)
summary(test_hbf)
plot(test_hbf)
autocorr.plot(test_hbf)To analyze the convergence we can observe normality in auto-correlation plots. If we wee substantial auto-correlation (lag > X), we can repeat the the MCMC for 100,000 simulations and sample every X steps by using thin = X in coda.samples(); where X is how often order occurs in the plot.
test_hbf <- coda.samples(model_hbf, c("par.b", "par.h", "par.m","par.p"), n.iter = 1e+05, thin = 30)
Depending on which hypothesis we are testing we could also eliminate b.1 or b.2. Or in other situations reparameterizations can reduce correlation.
ANOVA Example
In this example let's consider a study of 5 different treatment groups assigned to wear shirts with differing levels of cotton (15%, 20%, 25%, 30%, and 35%) and strength was measured. We'll code these levels as dummy variables and
Note that since 15% is the reference group we keep it as a constant.
model.1 <- "model {
### data model
for(i in 1:N){
y[i] ~dnorm(mu[i], tau)
mu[i] <- b.15 + b.20*lev.20[i] +b.25 *lev.25[i] +
b.30*lev.30[i] +b.35 * lev.35[i]
}
###
prior
b.15 ~dnorm(0,0.0001); ## referent group
b.20 ~dnorm(0,0.0001);
b.25 ~dnorm(0,0.0001);
b.30 ~dnorm(0,0.0001);
b.35 ~dnorm(0,0.0001);
tau ~dgamma(1,1)
### difference in strength between level 3 (25%) and level 4 (30%)
b.30.25 <- b.30-b.25
### estimated strength in groups (30%)
strength[1] <- b.15
strength[2] <- strength[1]+b.20
strength[3] <- strength[1]+b.25
strength[4] <- strength[1]+b.30
strength[5] <- strength[1]+b.35
}"ANCOVA Example
These are models that include a continuous covariate and a categorical variable with 2 categories.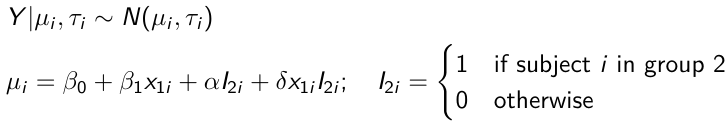
When the slope differs in the 2 groups and the lines are not parallel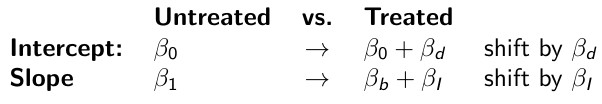
model.1 <- "model{
### data model
for(i in 1:N){
hbf_after[i] ~dlnorm(mu[i],tau)
Lhbf_baseline[i] <- log(hbf_baseline[i])
mu[i] <- beta.0 + beta.d*Drug[i] +
beta.b*(Lhbf_baseline[i]-mean(Lhbf_baseline[])) +
beta.i*Drug[i] *(Lhbf_baseline[i]-mean(Lhbf_baseline[]))
}
### prior density
beta.0 ~ dnorm(0,0.0001)
beta.d ~dnorm(0, 0.0001)
beta.b ~dnorm(0, 0.0001)
beta.i ~dnorm(0,0.0001)
tau ~ dgamma(1,1);
### inference
parameter[1] <- beta.0
parameter[2] <- beta.d
parameter[3] <- beta.b
parameter[4] <- beta.i
}"
### generate data
Drug = rep(0, nrow(hbf.data))
Drug[treatment == "Hy"] <- 1
table(Drug, hbf.data$Drug)
data.1 <- list(N = as.numeric(nrow(hbf.data)), hbf_baseline = hbf_baseline, hbf_after = hbf_after, Drug = Drug)
model_mean <- jags.model(textConnection(model.1), data = data.1,n.adapt = 1000)
update(model_mean, 10000)
test_mean <- coda.samples(model_mean, c("parameter"), n.iter = 10000)So if the interaction term is significant then we would conclude the treatment has an effect
Missing Values
Treat missing values in the response as unknown parameters and JAGS will generate them as a form of imputation. Missing data in the covariates however is not so easy.
Model Selection: DIC
- Model selection based on marginal likelihood is most robust but difficult to implement
- Often model search over many models is based on BIC using posterior estimates of parameters
- Model selection for a small number of models is based on posterior intervals
Start from the deviance: -2log(P(y | β, τ ))
Deviance information Criterion:
DIC = Dbar + pD = Dhat + 2 * pD
Dbar: -2 E(log(P(y | β, τ ))) = posterior mean of the deviance
Dhat: -2 log(P(y | β^, τ^ ) = point estimate of the deviance using the posterior means of the parameters
pD: Dbar - Dhat = Effective number of parameters
The model with the smallest DIC is estimated to be the model that would best predict a replicate dataset of the same structure as the observed.
No Comments