Sequencing Data and Analysis of Rare Variants
Genotyping arrays can be obtained at pre-selected sites for each sample. Ex. Genotyping sites known to be polymorphic based on prior sequencing.
Sequencing is obtaining "every" base in the exome or genome for each sample. Most of the sequence is identical across samples. This is used to find locations that are polymorphic, or differ across samples.
Whole Genome Sequencing
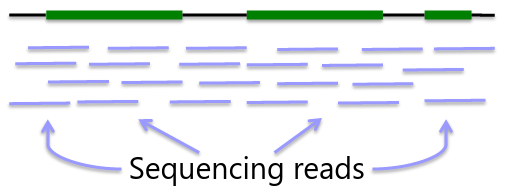
Genome: ~3GB per individual
Advantage - whole genome coverage
Disadvantage - cost ~$1000 for 30x, limited interpretability
Whole "Exome" Sequencing
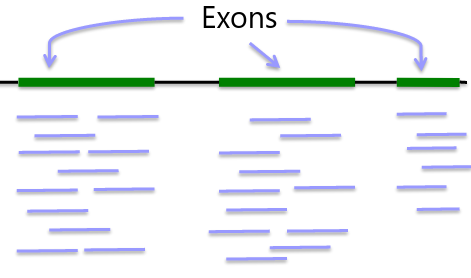
Exome: ~33MB per individual
Advantage: Covers protein coding regions, interpretable variation, cost ~$500
Disadvantage: Missing 99% genome coverage
VCF Format
Variant Calling File (VCF) is a standard format for storing sequencing data. It includes genotypes for all sites where at least one individual had alleles different from references alleles for all individuals in the study.
Every VCF file has three parts in the following order:
- Meta-information lines or Header (begins with ##)
- Last Meta-information line: Record Header (line beginning with #CHROM)
- Data lines (Body) contain marker and genotype data (one variant per line)
- A data line is called a VCF record
- Each VCF record has the same number of tab-seperated fields as the record header line
- The symbol "." is used to denote missing data

The first nine columns of the data record give information about the variants:
1. CHROM – the chromosome number/id
2. POS – the genome coordinate of the first base in the variant.
Within a chromosome, VCF records are sorted in order of increasing position.
3. ID – a semicolon-separated list of marker identifiers (often rsid)
4. REF – the reference allele expressed as a sequence of one or more A/C/G/T nucleotides (e.g. "A" or "AAC")
5. ALT – the alternate allele expressed as a sequence of one or more A/C/G/T nucleotides (e.g. "A" or "AAC"). If there is more than one alternate allele, the field is a comma-separated list of all alternate alleles.
6. QUAL - The Phred-scaled probability that a REF/ALT polymorphism exists at this site given sequencing data. A value of 10 indicates a 1 in 10^1 chance of error, 50 indicates 10^5 chance of error, etc.![]()
7. FILTER - Either "PASS" or a semicolon-separated list of failed quality control filters
8. INFO - Values in INFO are defined in the header. Contains additional information about the variant represented as tag-value pairs, where the tag and value are separated by an equals sign, and pairs are separated by colons. Usually it is information summarized from the samples, but can also include information from other sources such as population frequencies from a database.
9. FORMAT - Explanation of information in FORMAT are defined in the header. As for INFO, a colon-separated list. Describes the format of the data reported for each sample in the file.
There are many tools that can be used with VCF and BCF (binary version of VCF), such as bcftools and plink.
"Missing Heritability"
GWA studies are great for identifying SNP associations, but usually loci identified have small effects on traits. Much of the phenotypic variation or risk due to genetics ("heritability") is unexplained.
Unexplained variance/risk may be due to:
- Many additional variants with very small effects
- Non-SNP variants such as structural variants not typed by current GWAS chips.
- Epigenetic factors: methylation
- Over-estimation of heritability due to:
- Genetic interaction
- Dominance effects
- Shared environment
- Rare variants that we have have not yet been tested
Rare Variants
There are a lot of different rare variants, they are very common! Rare variants are more likely to be functional/deleterious. The best way to find rare variants is to sequence.
Recent expansion of the human population supports the theory of causal rare variants (there's 8 billion people today). Multiple causal rare variants have been found within genes. Animal studies suggest larger effects of rarer variants.
MAF (Minor Allele Frequency) cutoff can be used to define a "rare" variant (this level makes a difference in analysis).
Association Analysis with Rare Variants
The problem with single variant tests such as regression methods is that there are too few observations to provide a stable test.
We can combine rare variants by grouping them by gene region or functional information (exomes, non-synonymous or nonsense, predicted function), which can improve power.
Burden Tests
- Instead of testing rare variants individually, group variants likely to have similar function
- Score presence or absence of rare variants per individual -- use rare variant score to predict trait values
- Makes the assumption that the effect of all rare variants is in the same direction
- If all variants grouped in a test are causal, leads to a large increase in power
- Success (power) depends on:
- Number of associated variants contributing to signal
- Number of neutral (null) variants
- direct of effects
CAST
- Compare the frequency of cases and controls with at least one mutation within a gene or region
- Can be thought of as an indicator approach
- Xi = 1 when subject i has at least one rare variant in the region/gene
- Xi = 0 when a subject i has no rare variants
- CAST score can be tested against any type of outcome in regression framework
- Assumes same effect whether a single or multiple rare variants occur in a region in an individual
Unweighted Sum / CMC
- An extension of CAST
- An unweighted sum of the rare variants in a gene or region
- Commonly used to collapse rare variants into a score of minor alleles
- Regression (linear or logistic) used to test association with the rare variant score (sum)
- All variants included in the sum are assumed to have the same magnitude and direction of effect
RVT1
- An extension of CAST/CMC
- Instead of the sum, uses the proportion of sites at which an individual carries a rare variant as the predictor
- Accounts for missing data:
- ni - Number of rare variants for which subject i has been genotyped
- ri - Number out of the ni sites where individual i carries at least one rare allele
- Score is ri / ni