Sequencing Data and Analysis of Rare Variants
Genotyping arrays can be obtained at pre-selected sites for each sample. Ex. Genotyping sites known to be polymorphic based on prior sequencing.
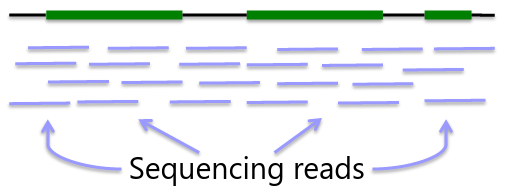
Sequencing is obtaining "every" base in the exome or genome for each sample. Most of the sequence is identical across samples. This is used to find locations that are polymorphic, or differ across samples.
Whole Genome Sequencing
Genome: ~3GB per individual
Advantage - whole genome coverage
Disadvantage - cost ~$1000 for 30x, limited interpretability
Whole "Exome" Sequencing
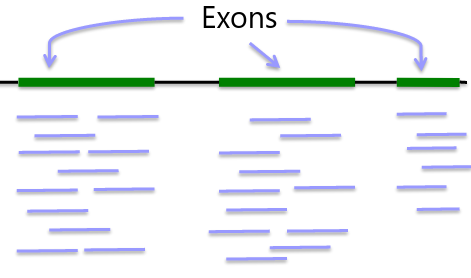
Exome: ~33MB per individual
Advantage: Covers protein coding regions, interpretable variation, cost ~$500
Disadvantage: Missing 99% genome coverage
VCF Format
Variant Calling File (VCF) is a standard format for storing sequencing data. It includes genotypes for all sites where at least one individual had alleles different from references alleles for all individuals in the study.
Every VCF file has three parts in the following order:
- Meta-information lines or Header (begins with ##)
- Last Meta-information line: Record Header (line beginning with #CHROM)
- Data lines (Body) contain marker and genotype data (one variant per line)
- A data line is called a VCF record
- Each VCF record has the same number of tab-seperated fields as the record header line
- The symbol "." is used to denote missing data

The first nine columns of the data record give information about the variants:
1. CHROM – the chromosome number/id
2. POS – the genome coordinate of the first base in the variant.
Within a chromosome, VCF records are sorted in order of increasing position.
3. ID – a semicolon-separated list of marker identifiers (often rsid)
4. REF – the reference allele expressed as a sequence of one or more A/C/G/T nucleotides (e.g. "A" or "AAC")
5. ALT – the alternate allele expressed as a sequence of one or more A/C/G/T nucleotides (e.g. "A" or "AAC"). If there is more than one alternate allele, the field is a comma-separated list of all alternate alleles.
6. QUAL - The Phred-scaled probability that a REF/ALT polymorphism exists at this site given sequencing data. A value of 10 indicates a 1 in 10^1 chance of error, 50 indicates 10^5 chance of error, etc.![]()
7. FILTER - Either "PASS" or a semicolon-separated list of failed quality control filters
8. INFO - Values in INFO are defined in the header. Contains additional information about the variant represented as tag-value pairs, where the tag and value are separated by an equals sign, and pairs are separated by colons. Usually it is information summarized from the samples, but can also include information from other sources such as population frequencies from a database.
9. FORMAT - Explanation of information in FORMAT are defined in the header. As for INFO, a colon-separated list. Describes the format of the data reported for each sample in the file.