Power and Sample Size Calculations for Association Studies
Review of Errors:errors and difference in means/proportions: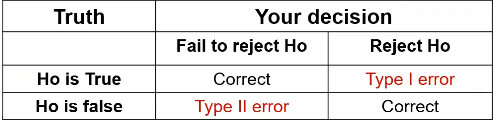
Type II error is represented by beta and type I error as alpha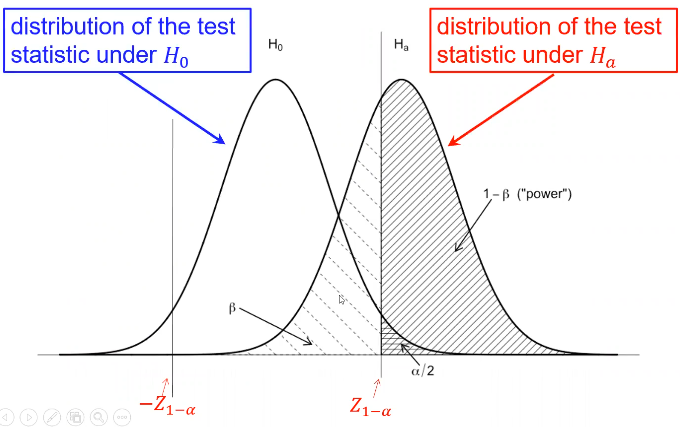
Where Z1 - (alpha /2)is the Z-value that creates the target value of alpha in the tail of the distribution. Power decreases as alpha increases.
Power also has a direct relationship with sample size. Sometimes we choose sample sized based on the desired power for a specific alternative hypothesis of interest, and other times we used a fixed sample size and significance level.
A Z-test of differences in proportions with 2 groups with different sample sizes: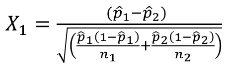
C is the mean of the distribution under the alternative hypothesis: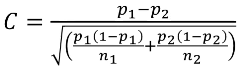
And the power can be calculated by taking the cdf of C minus the one sided Z-value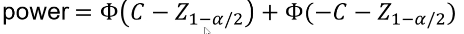
Likewise, we can use a similar z-test for differences in the mean of a quantitative trait: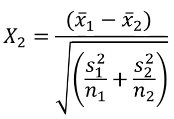
And the power can be calculated the same way as proportions.
Applying Power to Genetic Associations
- Difference in proportions: we can compute the power for a test that compares the allele or genotype frequency between cases and controls
- Difference in means: we can compare the mean trait values between individuals with different genotypes at a marker (and we can extend this to an F-test for a linear regression)
Dichotomous Traits
- Determine the expected genotype frequencies in cases and controls under the alternative hypothesis of interest
- Estimate power for given sample size or estimate minimum sample size required for given power and significance level under the planner analysis model
Genetic Model
- We usually use 4 parameters to specify the disease model for a dichotomous trait:
- Genotype relative risks y1 and y2
- Population prevalence of disease K
- Risk allele frequency q (the allele that increases risk of disease)
- To compute power, we need the genotype frequencies for cases and controls under the alternative hypothesis
- P(personal carries i risk alleles | affected)
- P(Person carries i risk alleles | unaffected)
- for i = 0,1, and 2
Genotype Relative Risk
Another way to parameterize penetrance, the probability that someone is affected given their genotype.
Penetrance fi = P(affected | i risk alleles) for i = 0,1, or 2 risk alleles at a locus
- f = (f0, f1, f2) is the penetrance for the genetic variant.
- f0 = f1 = f2 if there is no difference in probability of being affected by genotype -> the genotypes at this variant are not associated with case status
- If at least one fi != fj -> the genotype at this variant are associated with case status